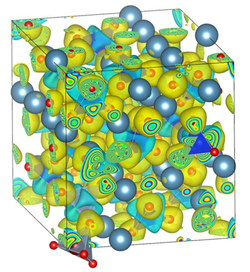
 The research applies density functional theory (DFT), to understand the role of defects on the structure and reactivity of complex oxides, with special applications to portland cement. Insights obtained from DFT simulations are expected to guide and inform new green-synthesis routes, to produce highly efficient cement chemistries. The DFT simulations link to experimental assessments of the water sensitivity of cement minerals.
The research applies density functional theory (DFT), to understand the role of defects on the structure and reactivity of complex oxides, with special applications to portland cement. Insights obtained from DFT simulations are expected to guide and inform new green-synthesis routes, to produce highly efficient cement chemistries. The DFT simulations link to experimental assessments of the water sensitivity of cement minerals.